Modeling Prion diseases in a dish
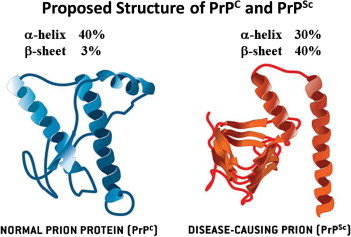
Neurodegenerative diseases are primarily characterized by the formation and aggregation of misfolded protein in the central nervous system. Prion diseases; a group of rare and fatal brain disease is caused by misfolding of cellular prion aggregates called scrapie prion proteins (PrP) or proteinaceous infectious particles (1). Pathological manifestations of prion disease include neuronal loss, activations of microglia and astrocytes, spongiform alterations, and formation of PrP aggregates. The pathogenicity of prion disease remains poorly understood, as the disease is usually diagnosed once at an advanced stage (2).
iPSC derived neurospheroids
iPSC based 3D cell cultures assay has been a major avenue to address the fundamental questions with regards to the pathogenicity of prion diseases, and subsequently produce a range of high throughput platforms for investigating possible treatments. The first eligible model was murine cerebellar organotypic slices, which consisted of increased PrP, upon exposure to prions (3). From these initial attempts more advanced models derived from murine neural stem cells were developed that includes 3D neurospheroids. The neurospheroids were highly effective, as it produced a mature culture within in 10 days and remained viable up to one month. Additionally prions were able to spread and cause toxic alterations to the neurospheroids during growth and differentiation, allowing for effective study of disease progression (4).
Human iPSC derived cerebral organoids
Human iPSC derived cerebral organoids are also used as a viable 3D model to study the effect of genetic predisposition on the etiology of prion diseases. The organoids were effective at mimicking astrogliosis, which is a typical occurrence in prion disease, but were unable to replicate aggregation of PrP as well as infection spread upon exposure to human PrP (5). Despite their drawbacks, these advanced 3D culture systems, derived from iPSCs, continue to provide insights in to the pathological events triggered by different human prion subtypes, and currently serve as a reliable platform for drug screens.
References
1. Scheckel, C., and Aguzzi, A. (2018). Prions, prionoids and protein misfolding disorders. Nat. Rev. Genet. 19, 405–418.
2. Knight, R. (2008). Clinical features and diagnosis of human prion diseases. Future Neurol. 3, 473–481.
3. Falsig, J., and Aguzzi, A. (2008). The prion organotypic slice culture assay–POSCA. Nat. Protoc. 3, 555–562.
4. Collins, S. J., and Haigh, C. L. (2017). Simplified murine 3D neuronal cultures for investigating neuronal activity and neurodegeneration. Cell Biochem. Biophys. 75, 3–13.
5. Matamoros-Angles, A., Gayosso, L. M., Richaud-Patin, Y., Di Domenico, A., Vergara, C., Hervera, A., et al. (2018). iPS cell cultures from a Gerstmann-Straussler-Scheinker Patient with the Y218N PRNP mutation recapitulate tau pathology. Mol. Neurobiol. 55, 3033–3048.
6. Lee, Jeongmin & Kim, Su & Jam, Kyu & Ju, Young & Woo, Hee-Jong. (2013). Prion Diseases as Transmissible Zoonotic Diseases. Osong public health and research perspectives. 4. 57-66.